Hybridization and Speciation | Natural History Museum Stuttgart

Research Publications
Explore our interdisciplinary research contributions spanning genetics, ecology, evolutionary biology, and conservation. Our publications cover topics such as hybridization, speciation mechanisms, population genomics, and ecological adaptations, reflecting our commitment to advancing understanding in these fields.
2024
The Role of Hybridization in Species Formation and Persistence. Cold Spring Harbor Perspectives in Speciation. 2024. Joshua V. Peñalba,..., and Ricardo J. Pereira
Hybridization, or interbreeding between different taxa, was traditionally considered to be rare and to have a largely detrimental impact on biodiversity, sometimes leading to the breakdown of reproductive isolation and even to the reversal of speciation. However, modern genomic and analytical methods have shown that hybridization is common in some of the most diverse clades across the tree of life, sometimes leading to rapid increase of phenotypic variability, to introgression of adaptive alleles, to the formation of hybrid species, and even to entire species radiations. In this review, we identify consensus among diverse research programs to show how the field has progressed. Hybridization is a multifaceted evolutionary process that can strongly influence species formation and facilitate adaptation and persistence of species in a rapidly changing world. Progress on testing this hypothesis will require cooperation among different subdisciplines.
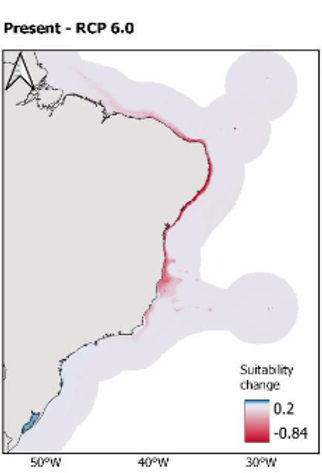
Larval dispersal and climate models provide insights into present and future distribution of a tropical sardine. Marine Biology Research. 2024. Coelho, J. F. R., Angeles-Gonzalez, L. E., Cahuich-López, M., Mariño-Tapia, I., & Queiroz Lima, S. M.
Climate change impacts the distribution of marine organisms and threatens fisheries. Marine protected areas (MPAs) may buffer the detrimental effects of environmental change by acting as biodiversity spillover to neighboring areas. Yet, it is uncertain whether MPAs on islands favor passive dispersal of species through oceanic currents, and how fish abundance and distribution will be impacted by ongoing global warming. Using the Brazilian-endemic scaled-sardines (Harengula sp.) as model, we implemented a Lagrangian particle-tracking model to estimate dispersal in the Brazilian coast and four MPAs in islands. Then, we projected an ecological niche model (ENM) of Harengula sp. to three climatic scenarios in 2100. Dispersal models suggested that three MPAs in islands export eggs and larvae acting as source of biomass to the coast. ENMs indicated a decrease in environmental suitability for Harengula sp. at the Equatorial Brazilian coast and a southward shift that increases as the climatic scenario is aggravated. Ocean warming may lead to a decrease in suitability for Harengula sp. in the northernmost part of its current distribution concomitant with an increase in offshore zones in the south of its current distribution. Ecosystem disturbance with environmental suitability shifts can aggravate the isolation of populations in islands.
Integration of genomic and ecological methods inform management of an undescribed, yet highly exploited, sardine species. Proceedings of the Royal Society B. 2024. Jéssica F. R. Coelho,..., Julia T. Verba, and Ricardo J. Pereira
Assessing genetic diversity within species is key for conservation strategies in the context of human-induced biotic changes. This is important in marine systems, where many species remain undescribed while being overfished, and conflicts between resource-users and conservation agencies are common. Combining niche modelling with population genomics can contribute to resolving those conflicts by identifying management units and understanding how past climatic cycles resulted in current patterns of genetic diversity. We addressed these issues on an undescribed but already overexploited species of sardine of the genus Harengula. We find that the species distribution is determined by salinity and depth, with a continuous distribution along the Brazilian mainland and two disconnected oceanic archipelagos. Genomic data indicate that such biogeographic barriers are associated with two divergent intraspecific lineages. Changes in habitat availability during the last glacial cycle led to different demographic histories among stocks. One coastal population experienced a 3.6-fold expansion, whereas an island-associated population contracted 3-fold, relative to the size of the ancestral population. Our results indicate that the island population should be managed separately from the coastal population, and that a Marine Protected Area covering part of the island population distribution can support the viability of this lineage
2023

Low population genetic structure is consistent with high habitat connectivity in a commercially important fish species (Lutjanus jocu). 2023. Marine Biology. Julia Tovar Verba,..., and Ricardo J. Pereira
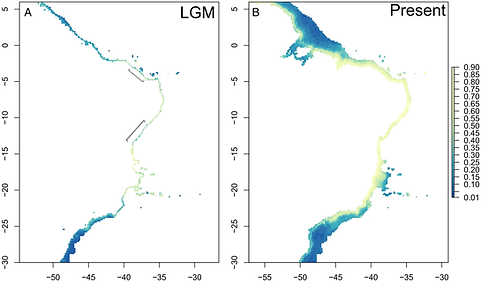
The level of habitat availability influences genetic divergence among populations and the genetic diversity within populations. In the marine environment, near-shore species are among the most sensitive to habitat changes. Knowledge of how historical environmental change affected habitat availability and genetic variation can be applied to the development of proactive management strategies of exploited species. Here, we modeled the contemporary and historical distribution of Lutjanus jocu in Brazil. We describe patterns of genomic diversity to better understand how climatic cycles might correlate with the species demographic history and current genetic structure. We show that during the Last Glacial Maximum, there were ecological barriers that are absent today, possibly dividing the range of the species into three geographically separated areas of suitable habitat. Consistent with a historical reduction in habitat area, our analysis of demographic changes shows that L. jocu experienced a severe bottleneck followed by a population size expansion. We also found an absence of genetic structure and similar levels of genetic diversity throughout the sampled range of the species. Collectively, our results suggest that habitat availability changes have not obviously influenced contemporary levels of genetic divergence between populations. However, our demographic analyses suggest that the high sensitivity of this species to environmental change should be taken into consideration for management strategies. Furthermore, the general low levels of genetic structure and inference of high gene flow suggest that L. jocu likely constitutes a single stock in Brazilian waters and, therefore, requires coordinated legislation and management across its distribution.
A Phylogenomic Assessment of Processes Underpinning Convergent Evolution in Open-Habitat Chats. 2023. Molecular Biology and Evolution. Niloofar A. Kakhki et al.
Insights into the processes underpinning convergent evolution advance our understanding of the contributions of ancestral, introgressed, and novel genetic variation to phenotypic evolution. Phylogenomic analyses characterizing genome-wide gene tree heterogeneity can provide first clues about the extent of ILS and of introgression and thereby into the potential of these processes or (in their absence) the need to invoke novel mutations to underpin convergent evolution. Here, we were interested in understanding the processes involved in convergent evolution in open-habitat chats (wheatears of the genus Oenanthe and their relatives). To this end, based on whole-genome resequencing data from 50 taxa of 44 species, we established the species tree, characterized gene tree heterogeneity, and investigated the footprints of ILS and introgression within the latter. The species tree corroborates the pattern of abundant convergent evolution, especially in wheatears. The high levels of gene tree heterogeneity in wheatears are explained by ILS alone only for 30% of internal branches. For multiple branches with high gene tree heterogeneity, D-statistics and phylogenetic networks identified footprints of introgression. Finally, long branches without extensive ILS between clades sporting similar phenotypes provide suggestive evidence for the role of novel mutations in the evolution of these phenotypes. Together, our results suggest that convergent evolution in open-habitat chats involved diverse processes and highlight that phenotypic diversification is often complex and best depicted as a network of interacting lineages.
An annotated chromosome-scale reference genome for Eastern Black-eared Wheatear (Oenanthe melanoleuca). G3, Genes Genomes Genetics, 2023. Valentina Peona,...,and Niloo A. Kakhki et al.
Pervasive convergent evolution and in part high incidences of hybridization distinguish wheatears (songbirds of the genus Oenanthe) as a versatile system to address questions at the forefront of research on the molecular bases of phenotypic and species diversification. To prepare the genomic resources for this venture, we here generated and annotated a chromosome-scale assembly of the Eastern black-eared wheatear (Oenanthe melanoleuca). This species is part of the Oenanthe hispanica complex that is characterized by convergent evolution of plumage coloration and high rates of hybridization. The long-read-based male nuclear genome assembly comprises 1.04 Gb in 32 autosomes, the Z chromosome, and the mitogenome. The assembly is highly contiguous (contig N50, 12.6 Mb; scaffold N50, 70 Mb), with 96% of the genome assembled at the chromosome level and 95.5% benchmarking universal single-copy orthologs (BUSCO) completeness. The nuclear genome was annotated with 18,143 protein-coding genes and 31,333 mRNAs (annotation BUSCO completeness, 98.0%), and about 10% of the genome consists of repetitive DNA. The annotated chromosome-scale reference genome of Eastern black-eared wheatear provides a crucial resource for research into the genomics of adaptation and speciation in an intriguing group of passerines.
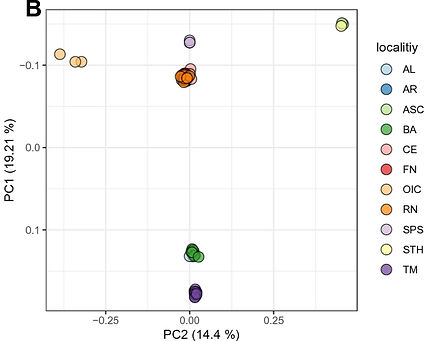
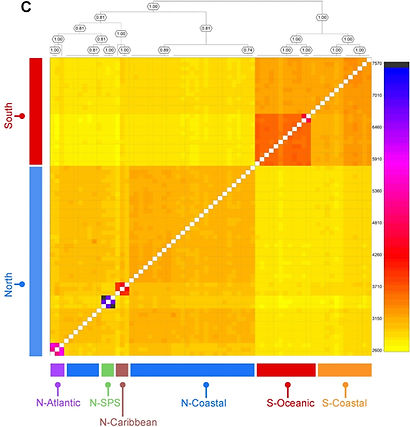
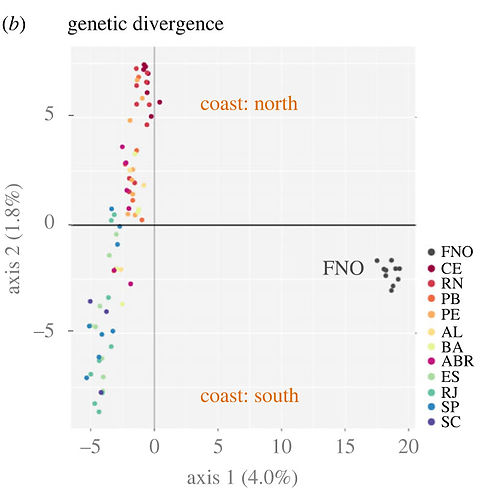
Population genomics of an Octopus species identify oceanographic barriers and inbreeding patterns. Marine Biology. 2023. Bernhard Bein,..., and Ricardo Pereira
Coastal marine ecosystems are highly productive and important for global fisheries. To mitigate over exploitation and to establish efficient conservation management plans for species of economic interest, it is necessary to identify the oceanographic barriers that condition divergence and gene flow between populations with those species, and that determine their relative amounts of genetic variability. Here, we present the first population genomic study of an Octopus species, Octopus insularis, which was described in 2008 and is distributed in coastal and oceanic island habitats in the tropical Atlantic Ocean. Using genomic data, we identify the South Equatorial current as the main barrier to gene flow between southern and northern parts of the range, followed by discontinuities in the habitat associated with depth. We find that genetic diversity of insular populations significantly decreases after colonization from the continental shelf, also reflecting low habitat availability. Using demographic modelling, we find signatures of a stronger population expansion for coastal relative to insular populations, consistent with estimated increases in habitat availability since the Last Glacial Maximum. The direction of gene flow is coincident with unidirectional currents and bidirectional eddies between otherwise isolated populations. Together, our results show that oceanic currents and habitat breaks are determinant in the diversification of coastal marine species where adults have a sedentary behavior but paralarvae are dispersed passively, shaping standing genetic variability within populations. Lower genetic diversity within insular populations implies that these are particularly vulnerable to current human exploitation and selective pressures, calling for the revision of their protection status.
2022
Genome-wide evidence supports mitochondrial relationships and pervasive parallel phenotypic evolution in open-habitat chats. Molecular Phylogenetics and Evolution. 2022. Manuel Schweizer,..., and Niloo A. Kakhki et al.
In wheatears and related species (‘open-habitat chats’), molecular phylogenetics has led to a comprehensively revised understanding of species relationships and species diversity. Phylogenetic analyses have suggested that, in many cases, phenotypic similarities do not reflect species’ relationships, revealing traditionally defined genera as non-monophyletic. This led to the suggestion of pervasive parallel evolution of open-habitat chats’ plumage coloration and ecological phenotypes. However, to date, the molecular evidence for the phylogenetic relationships among open-habitat chats is mainly limited to mitochondrial DNA. Here, we assessed whether the mitochondrial relationships are supported by genome-wide data. To this end, we reconstructed the species tree among 14 open-habitat chat taxa using multi-species coalescent analyses based on ~1’300 SNPs. Our results confirm previous ones based chiefly on mitochondrial DNA; notably the paraphyly of the Oenanthe lugens complex and the clustering of individual species formerly placed in the genera Cercomela and Myrmecocichla within Oenanthe. Since several variable morphological and ecological characteristics occur in multiple places across the open-habitat chat phylogeny, our study consolidates the evidence for pervasive parallel evolution in the plumage coloration and ecology of open-habitat chats.
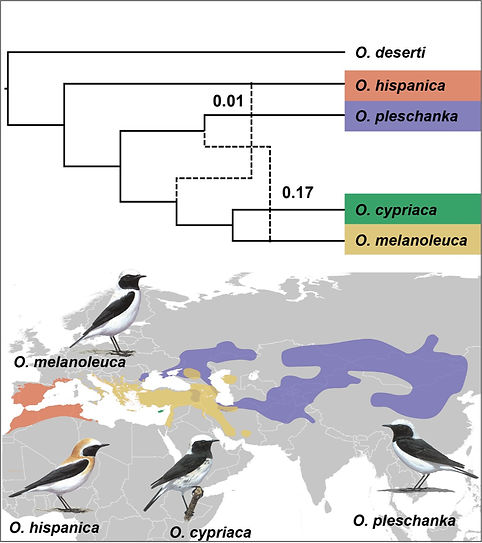
Parallel plumage color evolution and introgressive hybridization in wheatears. Journal of Evolutionary Biology. 2022. Manuel Schweizer,..., and Niloo A. Kakhki et al.
Genetic and phenotypic mosaics, in which various phenotypes and different genomic regions show discordant patterns of species or population divergence, offer unique opportunities to study the role of ancestral and introgressed genetic variation in phenotypic evolution. Here, we investigated the evolution of discordant phenotypic and genetic divergence in a monophyletic clade of four songbird taxa – pied wheatear (O. pleschanka), Cyprus wheatear (O. cypriaca), and western and eastern subspecies of black‐eared wheatear (O. h. hispanica and O. h. melanoleuca). Phenotypically, black back and neck‐sides distinguish pied and Cyprus wheatears from the white‐backed/necked black‐eared wheatears. Meanwhile, mitochondrial variation only distinguishes western black‐eared wheatear. In the absence of nuclear genetic data, and given frequent hybridization among eastern black‐eared and pied wheatear, it remains unclear whether introgression is responsible for discordance between mitochondrial divergence patterns and phenotypic similarities, or whether plumage coloration evolved in parallel. Multispecies coalescent analyses of about 20'000 SNPs obtained from RAD data mapped to a draft genome assembly resolve the species tree, provide evidence for the parallel evolution of color phenotypes, and establish western and eastern black‐eared wheatears as independent taxa that should be recognized as full species. The presence of the entire admixture spectrum in the Iranian hybrid zone and the detection of footprints of introgression from pied into eastern black‐eared wheatear beyond the hybrid zone despite strong geographic structure of ancestry proportions furthermore suggest a potential role for introgression in parallel plumage color evolution. Our results support the importance of standing heterospecific and/or ancestral variation in phenotypic evolution. This article is protected by copyright. All rights reserved.
Polymorphism-aware estimation of species trees and evolutionary forces from genomic sequences with RevBayes. Methods in Ecology and Evolution. 2022. Rui Borges,..., and Pereira et al.
The availability of population genomic data through new sequencing technologies gives unprecedented opportunities for estimating important evolutionary forces such as genetic drift, selection and mutation biases across organisms. Yet, analytical methods that can handle polymorphisms jointly with sequence divergence across species are rare and not easily accessible to empiricists.We implemented polymorphism-aware phylogenetic models (PoMos), an alternative approach for species tree estimation, in the Bayesian phylogenetic software RevBayes. PoMos naturally account for incomplete lineage sorting, which is known to cause difficulties for phylogenetic inference in species radiations, and scale well with genome-wide data. Simultaneously, PoMos can estimate mutation and selection biases.We have applied our methods to resolve the complex phylogenetic relationships of a young radiation of Chorthippus grasshoppers, based on coding sequences. In addition to establishing a well-supported species tree, we found a mutation bias favouring AT alleles and selection bias promoting the fixation of GC alleles, the latter consistent with GC-biased gene conversion. The selection bias is two orders of magnitude lower than genetic drift, validating the critical role of nearly neutral evolutionary processes in species radiation.PoMos offer a wide range of models to reconstruct phylogenies and can be easily combined with existing models in RevBayes—for example, relaxed clock and divergence time estimation—offering new insights into the evolutionary processes underlying molecular evolution and, ultimately, species diversification.
Out of Asia? Expansion of Eurasian Lyme borreliosis causing genospecies display unique evolutionary trajectories. Molecular Ecology. 2022. Robert E. Rollins,..., and Ricardo J. Pereira et al.
Vector-borne pathogens exist in obligate transmission cycles between vector and reservoir host species. Host and vector shifts can lead to geographic expansion of infectious agents and the emergence of new diseases in susceptible individuals. Three bacterial genospecies (Borrelia afzelii, Borrelia bavariensis, and Borrelia garinii) predominantly utilize two distinct tick species as vectors in Asia (Ixodes persulcatus) and Europe (Ixodes ricinus). Through these vectors, the bacteria can infect various vertebrate groups (e.g., rodents, birds) including humans where they cause Lyme borreliosis, the most common vector-borne disease in the Northern hemisphere. Yet, how and in which order the three Borrelia genospecies colonized each continent remains unclear including the evolutionary consequences of this geographic expansion. Here, by reconstructing the evolutionary history of 142 Eurasian isolates, we found evidence that the ancestors of each of the three genospecies probably have an Asian origin. Even so, each genospecies studied displayed a unique substructuring and evolutionary response to the colonization of Europe. The pattern of allele sharing between continents is consistent with the dispersal rate of the respective vertebrate hosts, supporting the concept that adaptation of Borrelia genospecies to the host is important for pathogen dispersal. Our results highlight that Eurasian Lyme borreliosis agents are all capable of geographic expansion with host association influencing their dispersal; further displaying the importance of host and vector association to the geographic expansion of vector-borne pathogens and potentially conditioning their capacity as emergent pathogens.
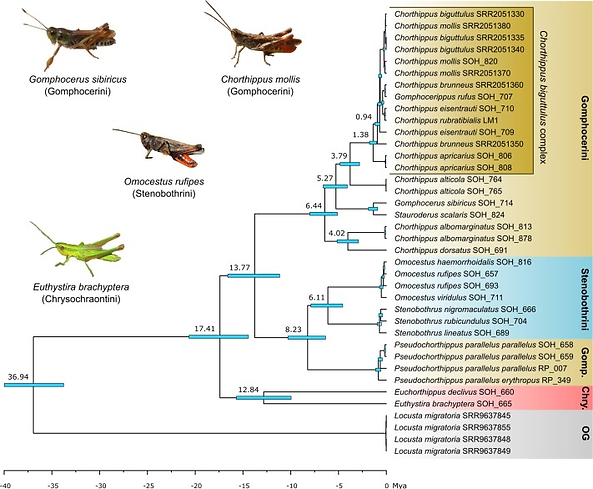
Transcriptomic data reveals nuclear-mitochondrial discordance in Gomphocerinae grasshoppers (Insecta: Orthoptera: Acrididae). Molecular Phylogenetics and Evolution. 2022. Oliver Hawlitschek,..., and Ricardo J. Pereira.
The phylogeny of many groups of Orthoptera remains poorly understood. Previous phylogenetic studies largely restricted to few mitochondrial markers found many species in the grasshopper subfamily Gomphocerinae to be para- or polyphyletic, presumably because of incomplete lineage sorting and ongoing hybridization between putatively young lineages. Resolving the phylogeny of the Chorthippus biguttulus species complex is important because many morphologically cryptic species occupy overlapping ranges across Eurasia and serve important ecological functions. We investigated whether multispecies coalescent analysis of 540 genes generated by transcriptome sequencing could resolve the phylogeny of the C. biguttulus complex and related Gomphocerinae species. Our divergence time estimates confirm that Gomphocerinae is a very young radiation, with an age estimated at 1.38 (2.35-0.77) mya for the C. biguttulus complex. Our estimated topology based on complete mitogenomes recovered some species as para- or polyphyletic. In contrast, the multispecies coalescent based on nuclear genes retrieved all species as monophyletic clusters, corroborating most taxonomic hypotheses. Our results underline the importance of using nuclear multispecies coalescent methods for studying young radiations and highlight the need of further taxonomic revision in Gomphocerinae grasshoppers.
Extensive introgression at late stages of species formation: Insights from grasshopper hybrid zones. Molecular Ecology. 2022. Linda Hagberg,..., and Ricardo J. Pereira.
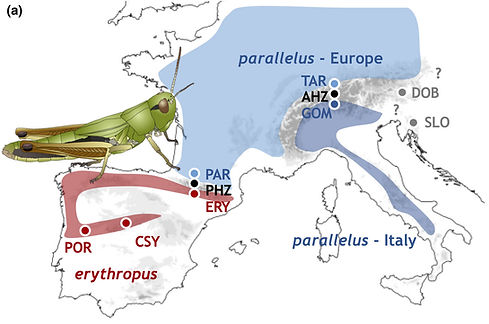
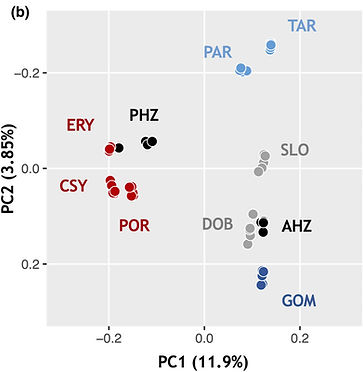
The process of species formation is characterized by the accumulation of multiple reproductive barriers. The evolution of hybrid male sterility, or Haldane’s rule, typically characterizes later stages of species formation, when reproductive isolation is strongest. Yet, understanding how quickly reproductive barriers evolve and their consequences for maintaining genetic boundaries between emerging species remains a challenging task because it requires studying taxa that hybridize in nature. Here, we address these questions using the meadow grasshopper Pseudochorthippus parallelus, where populations that show multiple reproductive barriers, including hybrid male sterility, hybridize in two natural hybrid zones. Using mitochondrial data, we infer that such populations diverged some 100,000 years ago, at the beginning of the last glacial cycle in Europe. Nuclear data show that contractions at multiple glacial refugia, and post-glacial expansions have facilitated genetic differentiation between lineages that today interact in hybrid zones. We find extensive introgression throughout the sampled species range, irrespective of the current strength of reproductive isolation. Populations exhibiting hybrid male sterility in two hybrid zones show repeatable patterns of genomic differentiation, consistent with shared genomic constraints affecting ancestral divergence or with the role of those regions in reproductive isolation. Together, our results suggest that reproductive barriers that characterize late stages of species formation can evolve relatively quickly, particularly when associated with strong demographic changes. Moreover, we show that such barriers persist in the face of extensive gene flow, allowing future studies to identify associated genomic regions.
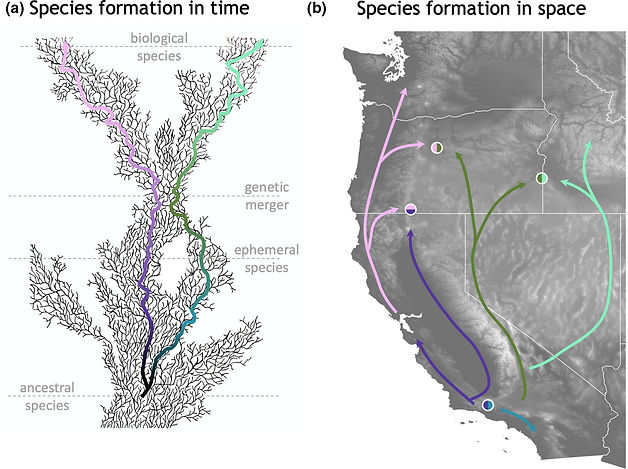
A lizard with two tales: What diversification within Sceloporus occidentalis teaches us about species formation. Molecular Ecology. 2022. Ricardo J. Pereira, and Sonal Singhal.
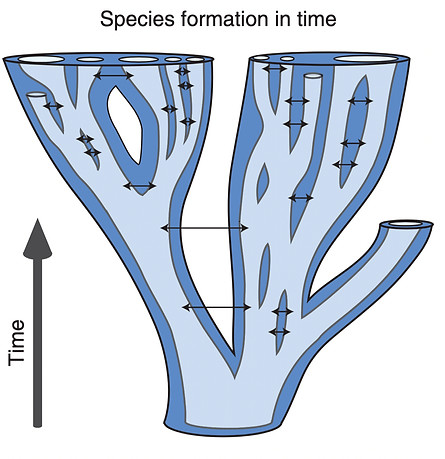
In 1859, Charles Darwin proposed that species are not fundamentally different from subspecies or the varieties from which they evolve. A century later, Dobzhansky (1958) suggested that many such lineages are ephemeral and are likely to reverse differentiation through introgression (Figure 1a); only a few evolve complete reproductive isolation and persist in sympatry. In this issue of Molecular Ecology, Bouzid et al. (2021) showed how new analytical methods, when applied to genomic data, allow us to more precisely determine whether or not species formation follows the paths outlined by Darwin and Dobzhansky (Figure 1b). The authors studied the diversification of the lizard Sceloporus occidentalis, finding a continuum of genetic interactions between the preservation of genetic identity to genetic merger, analogous to what is exemplified by ring species. In doing so, they teach us two tales about species formation: that lineages are fractal byproducts of evolutionary processes such as genetic drift and selection, and that lineages are often ephemeral and do not always progress into fully reproductively isolated taxa. Studying ephemeral lineages like those in S. occidentalis allows us to capture divergence at its earliest stages, and potentially to determine the factors that allow lineages to remain distinct despite pervasive gene flow. These lineages thus serve as a natural laboratory to address long standing hypotheses about species formation.